
Flexible genomic over-representation analysis
Castillo-Davis, C.I. and D.L. Hartl 2003. Bioinformatics 19(7):891-892 | Supp. Material.
What is GeneMerge?
GeneMerge tests for statistical over-representation of gene attributes in a given set of genes compared to the genome background.
It answers the question, Is there functional or categorical 'enrichment' of some kind among this set of genes?
GeneMerge is useful for the analysis of gene expression data like RNA-seq and other genome-scale data, for example, those generated by evolutionary and population genomic studies.
It uses the statistically exact hypergeometric probability distribution to calculate over-representation of gene attributes and provides Bonferroni and False Discovery Rate (FDR) correction for multiple tests using the method of Benjamini & Hochberg (1995).
Why Use GeneMerge?
The big advantage of GeneMerge over similar tools is that it is extremely flexible and it can be used with any type of gene-association data and with the genomes of non-model organisms.With it's simple to create gene-association file format you can analyze your genomic data for over-representation of any gene or locus-based attribute, not just Gene Ontology (GO) terms. GeneMerge has been used in many types of published studies with diverse species and gene-association data.
GeneMerge is an easy to use, Unix-inspired, command-line tool for investigators who seek to analyze their data in a powerful and flexible way.
Download
GeneMerge runs on any computer that has PERL installed. Perl comes with MacOS, Linux, and Unix, and can be easily installed on Windows. No external libraries or other software is needed. Since GeneMerge is a single self-contained program with no dependencies, it is easy to install, use, and incorporate into bioinformatic pipelines.
GeneMerge is free software; you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation.
MacOS / Unix / Linux
GeneMerge1.5.tar.gz
Windows
GeneMergeWIN-1.5.zip Note: you must install PERL first. ActivePerl has been tested and is recommended.
The above packages include monthly updated Gene Ontology files for all
GO species, including both specific and higher-order (all parent term) files in all three GO namespaces (molecular function,
biological process, and cellular component) as well as other types of gene-association files for select species. You can download these separately.
Documentation
The rationale and statistical methods behind GeneMerge can be found in Castillo-Davis & Hartl (2003). Details on program installation, usage options, and how to create your own gene-association files can be found in the GeneMerge Manual.
Publication
Castillo-Davis, C.I. and D.L. Hartl 2003. Bioinformatics 19(7):891-892Manual
GeneMerge1.5-Documentation.pdf
Quick Look
GeneMerge is called on the command-line as follows:
GeneMerge1.5.pl gene-association description population study output
Input files are simple text.
The study file contains a list of the gene names under investigation, the population file contains a list of all genes from which the study set was drawn (often a genome). The gene-association and description files contain gene-ID and ID-description pairs, respectively.
Output is tab-delimited text and html:
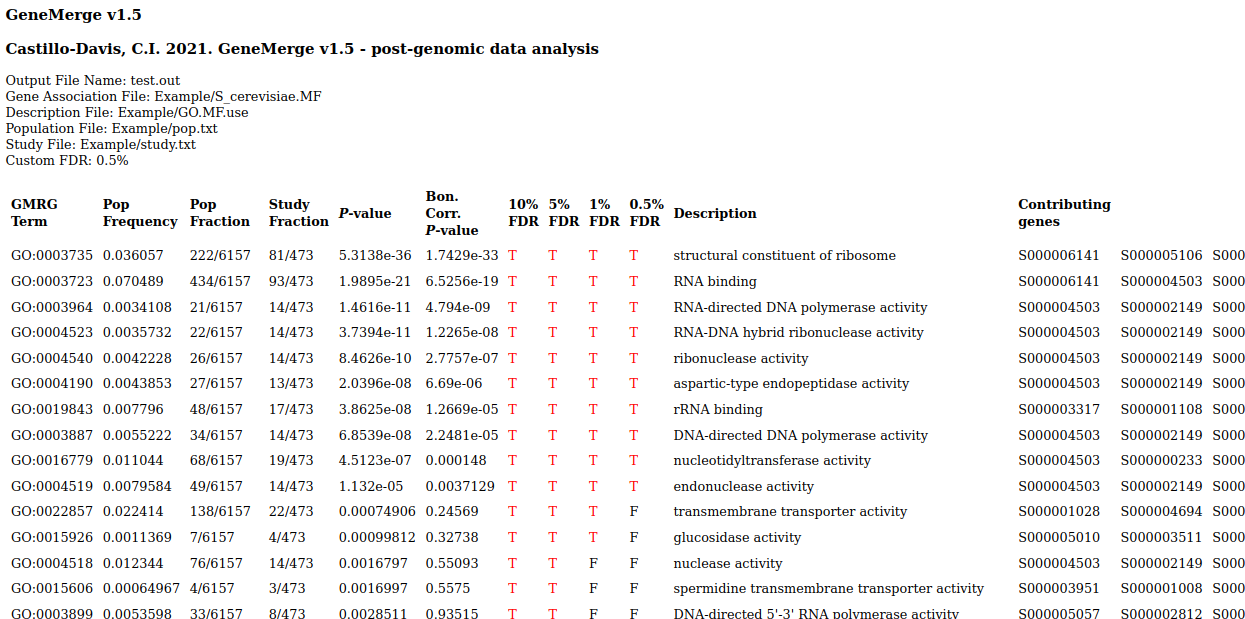 . . .
. . .
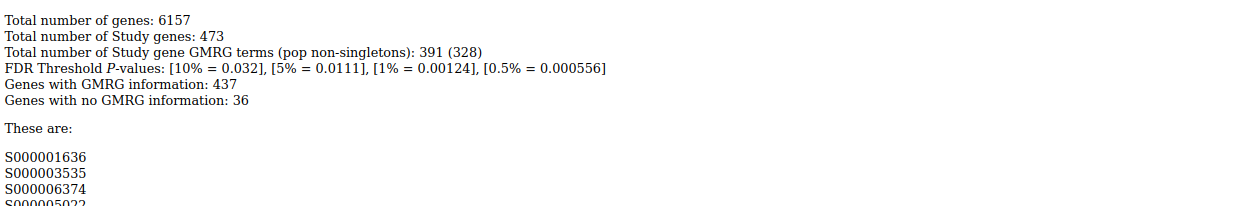
News
- GeneMerge version 1.5 is now available. It has been optimized to run
faster (10-80x) on large, well-annotated genomes and maintains all the same features and interfaces.
- GeneMerge v1.4 has been archived
for those interested in using it for study replication or other purposes.